

- Definition and Description of Smith Lemli Opitz Syndrome
- Signs and Symptoms of Smith Lemli Opitz Syndrome
- The Frequency or Prevalence of Smith Lemli Opitz Syndrome – Affected Populations
- The Causes of Smith Lemli Opitz Syndrome
- How is Smith Lemli Opitz Syndrome Transferred or Inherited from One Generation to Another or from Parents to Offspring?
- Screening and Diagnosis of Smith Lemli Opitz Syndrome
- Treatment and Management of Smith Lemli Opitz Syndrome
- Related Disorders Associated with Smith Lemli Opitz Syndrome
- Other Names for Smith Lemli Opitz Syndrome
- Conclusion
Definition and Description of Smith Lemli Opitz Syndrome
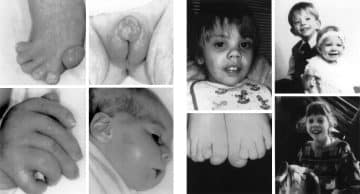
Smith-Lemli-Opitz syndrome is a congenital or developmental condition that affects many bodily systems. This disorder is distinguished by unusual facial characteristics, a tiny head shape (microcephaly), intellectual handicap or learning difficulties, and behavioral difficulties.
Many of the children impacted by this syndrome have autism, a developmental disorder that hampers speech and social interaction. Heart, lung, kidney, gastrointestinal system, and genitalia malformations are also prevalent.
Newborns having Smith Lemli Opitz syndrome have low muscle tone (hypotonia), have eating problems, and develop at a slower rate than typical infants. The majority of affected people have joined third and second toes (syndactyly), while others have extra toes and fingers (polydactyly).
Quick Facts About Smith Lemli Opitz Syndrome (SLOS)
| A | B |
|---|---|
| Definition | A genetic disorder affecting cholesterol metabolism, leading to multiple developmental issues |
| Also Known As | 7-dehydrocholesterol reductase deficiency, RSH syndrome, SLOS |
| Genetic Cause | Mutations in the DHCR7 gene, which is responsible for producing the enzyme 7-dehydrocholesterol reductase |
| Inheritance Pattern | Autosomal recessive, meaning both parents must carry one copy of the mutated gene |
| Symptoms | Distinctive facial features, small head size (microcephaly), intellectual disability or learning problems, behavioral problems, malformations of the heart, lungs, kidneys, gastrointestinal tract, and genitalia, weak muscle tone (hypotonia), feeding difficulties, slow growth, fused toes (syndactyly), extra fingers or toes (polydactyly) |
| Diagnosis | Based on clinical features, biochemical testing showing elevated 7-dehydrocholesterol levels, and genetic testing confirming DHCR7 mutations |
| Prevalence | Estimated to occur in 1 in 20,000 to 1 in 60,000 live births |
| Treatment | No cure; management focuses on symptomatic treatment and may include dietary cholesterol supplementation, behavioral therapies, and surgical interventions for physical anomalies |
| Treatment | No cure; management focuses on symptomatic treatment and may include dietary cholesterol supplementation, behavioral therapies, and surgical interventions for physical anomalies |
| Prognosis | Varies widely; some individuals have mild symptoms and lead relatively normal lives, while others have severe disabilities and health issues |
| Research | Ongoing to better understand the disease mechanisms, improve diagnostic methods, and develop targeted therapies |
| First Described | In 1964 by David Smith, Luc Lemli, and John Opitz. |
To learn more about this syndrome, you can watch the video Smith Lemli Opitz syndrome below;
Signs and Symptoms of Smith Lemli Opitz Syndrome
Smith Lemli Opitz syndrome manifests itself in a variety of ways. Mildly afflicted people may have modest physical anomalies as well as learning and behavioral issues. Severe instances may be fatal, including considerable intellectual incapacity and significant physical deformities.
The following are said to manifest as the signs and symptoms of Smith Lemli Opitz Syndrome:
- SLOS
- DHCR7 abnormality
- SLO syndrome
- 7-dehydrocholesterol reductase deficiency
- RSH syndrome
- Lethargy
- Obtundation or coma
- Respiratory failure
- Hearing loss
- Visual loss
- Vomiting
- Feeding difficulties
- Failure to thrive
- Constipation
- Cyanosis
- Congestive heart failure
- Photosensitivity
Neuropsychiatric and neurodevelopmental abnormalities are common and include variable intellectual disability (ID), aberrant behavior, and autism.
SLOS characteristics differ from patient to patient. Slow development before and after delivery, a tiny head (microcephaly), and a cleft in the mouth's roof are all common observations (cleft palate).
The majority of newborns have variations in their toes and/or fingers. This comprises fused toes as well as additional fingers or toes. Males suffering from SLOS may have undeveloped genitals that resemble female genitals. People with SLOS often have behavioral issues, sleep issues, and moderate to severe intellectual handicaps.
People with SLOS have distinct facial characteristics. Their eyes may seem droopy (ptosis). A fold in the interior corner of the eyes and wrinkles mostly on eyelids are also possible. SLOS children may have a tiny nose and nostril discrepancies (anteverted nares).
Other typical facial characteristics include variations in the top lip, a tiny jaw, and prominent ears. Cataracts are one kind of vision issue that may arise. There might also be variations in the spacing and/or the number of teeth.
Seizures, cardiac abnormalities, and decreased muscular tone are uncommon findings (hypotonia). SLOS may cause constriction at the upper part of the abdomen or stomach (pyloric stenosis) and bowel blockage (obstruction). Light sensitivity (photosensitivity) is also widespread.
The Frequency or Prevalence of Smith Lemli Opitz Syndrome – Affected Populations
Smith Lemli Opitz syndrome is thought to afflict 1 in 20,000 to 60,000 births. Whites of European descent, especially those from Central European nations like Slovakia and the Czech Republic, are most likely to have this illness.
It is quite uncommon among Asian and African populations. This illness affects both men and women equally. Females, on the other hand, are less likely to receive a diagnosis since they don't have any genital differences.
The Causes of Smith Lemli Opitz Syndrome
Smith Lemli Opitz syndrome is due to mutations (abnormal changes) in the DHCR7 gene1https://medlineplus.gov/genetics/condition/smith-lemli-opitz-syndrome/, which codes for the enzyme 7-dehydrocholesterol reductase. This enzyme is in charge of the last stage of cholesterol synthesis.
Cholesterol is a waxy, fat-like molecule generated in the body and taken from animal-derived meals (particularly fish, dairy products, poultry, meat, and egg yolks). Cholesterol is required for optimal embryonic development and serves a crucial role both before and after birth. It is a component of the cell membranes as well as the protective coating that covers nerve cells (myelin). Furthermore, cholesterol is involved in the creation of various hormones.
Mutations in the DHCR7 gene diminish or abolish 7-dehydrocholesterol reductase function, preventing cells from generating adequate cholesterol. In addition, the absence of this enzyme permits harmful byproducts of cholesterol manufacturing to accumulate in the bloodstream, nervous system, as well as other organs.
Low cholesterol levels combined with a buildup of other chemicals are likely to affect the development and growth of numerous physiological systems. However, it is unclear how each anomaly leads to the particular symptoms of Smith-Lemli-Opitz syndrome.
How is Smith Lemli Opitz Syndrome Transferred or Inherited from One Generation to Another or from Parents to Offspring?
This disorder is inheritable in an autosomal fashion, which means that both of the gene's copies in each cell are mutated. The parents of a person with such an autosomal recessive disorder each have one copy of the defective gene, although they usually do not have the condition.
Screening and Diagnosis of Smith Lemli Opitz Syndrome
Prenatally
The most characteristic biochemical indicator of SLOS is an increased concentration of 7-DHC (reduced cholesterol levels are also typical, but appear in other disorders as well). Thus, prenatally, SLOS is diagnosed upon finding an elevated 7-DHC: total sterol ratio in fetal tissues, or increased levels of 7-DHC in amniotic fluid.
The 7-DHC: total sterol ratio can be measured at 11–12 weeks of gestation by chorionic villus sampling, and elevated 7DHC in amniotic fluid can be measured by 13 weeks. Furthermore, if parental mutations are known, DNA testing of amniotic fluid or chorionic villus samples may be performed. Micrograph showing chorionic villi, the tissue that is collected with chorionic villus sampling and used to test for SLOS.
Amniocentesis (the process of sampling amniotic fluid) and chorionic villus sampling cannot be performed until approximately 3 months into the pregnancy. Given that SLOS is a very severe syndrome, parents may want to choose to terminate their pregnancy if their fetus is affected.
Amniocentesis and chorionic villus sampling leave very little time to make this decision (abortions become more difficult as the pregnancy advances), and can also pose severe risks to the mother and baby. Thus, there is a very large desire for noninvasive midgestational diagnostic tests. Examining the concentrations of sterols in maternal urine is one potential practice to identify SLOS prenatally.
During pregnancy, the fetus is solely responsible for synthesizing the cholesterol needed to produce estriol. A fetus with SLOS cannot produce cholesterol, and may use 7-DHC or 8-DHC as precursors for estriol instead. This creates 7- or 8-dehydrosteroids (such as 7-dehydroestriol), which may show up in the maternal urine.
These are novel metabolites due to the presence of a normally reduced double bond at carbon 7 (caused by the inactivity of DHCR7) and may be used as indicators of SLOS.
Other cholesterol derivatives which possess a double bond at the 7th or 8th position and are present in maternal urine may also be indicators of SLOS. 7- and 8-dehydropregnanetriols have been shown to be present in the urine of mothers with an affected fetus but not with an unaffected fetus, and thus are used in diagnosis.
These pregnatrienes originated in the fetus and traveled through the placenta before reaching the mother. Their excretion indicates that neither the placenta nor the maternal organs have the necessary enzymes needed to reduce the double bond of these novel metabolites.
Postnatally
If SLOS goes undetected until after birth, diagnosis may be based on the characteristic physical features as well as finding increased plasma levels of 7-DHC.2https://emedicine.medscape.com/article/949125-overview
There are many different ways of detecting 7-DHC levels in blood plasma, one way is using the Liebermann–Burchard (LB) reagent. This is a simple colorimetric assay developed with the intention of use for large-scale screening.
When treated with the LB reagent, SLOS samples turn pink immediately and gradually become blue; normal blood samples are initially colorless and develop a faint blue color. Although this method has limitations and is not used to give a definitive diagnosis, it has appeal in that it is a much faster method than using cell cultures.
Another way of detecting 7-DHC is through gas chromatography, a technique used to separate and analyze compounds. Selected ion monitoring gas chromatography/mass spectrometry (SIM-GC/MS) is a very sensitive version of gas chromatography and permits the detection of even mild cases of SLOS.
Other methods include time-of-flight mass spectrometry, particle-beam LC/MS, electrospray tandem MS, and ultraviolet absorbance, all of which may be used on either blood samples, amniotic fluid, or chorionic villus. Measuring levels of bile acids in patients' urine, or studying DCHR7 activity in tissue culture is also a common postnatal diagnostic technique.
SLOS is diagnosed based on physical observations as well as biochemical or genetic studies. Protein levels present in the blood are measured through biochemical tests. Protein 7-dehydrocholesterol is increased in SLOS patients. Genetic testing examines a patient's genes for alterations. SLOS is confirmed if the copies of the DHCR7 gene have detrimental alterations.
The results of a pregnancy test may indicate SLOS. To confirm the diagnosis, an amniocentesis might be done. During pregnancy, an amniocentesis utilizes a needle to extract a little quantity of fluid around the fetus. This fluid may be examined for 7-DHC levels and DHCR7 gene alterations.
Fetal ultrasonography may reveal anomalies suggestive of Smith-Lemli-Opitz syndrome. Confirmatory prenatal diagnostic testing is currently available by genetic mutation analysis. Postnatally, the syndrome is usually suspected clinically, but biochemical and/or genetic testing is necessary for diagnosis.
Plasma total cholesterol and/or low-density lipoprotein (LDL) cholesterol levels may be low but are not universally low. Measurement of plasma sterols, including, at a minimum, cholesterol, and 7DHC, is the biochemical test for Smith-Lemli-Opitz syndrome.
Treatment and Management of Smith Lemli Opitz Syndrome
Management of individuals with SLOS is complex and often requires a team of specialists. Some of the congenital malformations (cleft palate) can be corrected with surgery3https://en.wikipedia.org/wiki/Smith–Lemli–Opitz_syndrome#Treatment. Other treatments have yet to be proven successful in randomized studies, however, anecdotally they appear to cause improvements.
Treatment with Standard Therapies
SLOS treatment is tailored to the child's individual issues. Cleft palate, heart problems, and genital malformations may need surgery for certain children. The early educational intervention seems to be critical for children with cognitive and developmental difficulties.
Several of the symptoms and signs of SLOS, including growth, may be improved by eating foods high in cholesterol and using bile acid supplements. Statins, like Simvastatin, may lower DHC levels safely and may help with certain behavioral issues.
Experimental or Investigational Therapies
New therapy for SLOS is being researched. Cholic acid, antioxidants, cholesterol, and other dietary supplements are now being studied in clinical studies. There is also some indication that timely liver transplantation in persons with SLOS might be explored.
Cholesterol supplementation
Currently, the most common form of treatment for SLOS involves dietary cholesterol supplementation. Anecdotal reports indicate that this has some benefits; it may result in increased growth, lower irritability, improved sociability, less self-injurious behaviour, less tactile defensiveness, fewer infections, more muscle tone, less photosensitivity, and fewer autistic behaviours.
Cholesterol supplementation begins at a dose of 40–50 mg/kg/day, increasing as needed. It is administered either through consuming foods high in cholesterol (eggs, cream, liver), or as purified food-grade cholesterol.
Younger children and infants may require tube feeding. However, dietary cholesterol does not reduce the levels of 7DHC, cannot cross the blood-brain barrier, and does not appear to improve developmental outcomes.
One empirical study found that cholesterol supplementation did not improve developmental delay, regardless of the age at which it began. This is likely because most developmental delays stem from malformations of the brain, which dietary cholesterol cannot ameliorate due to its inability to cross the blood-brain barrier.
Simvastatin therapy
HMG-CoA reductase inhibitors have been examined as a treatment for SLOS. Given that this catalyzes the rate-limiting step in cholesterol synthesis, inhibiting it would reduce the buildup of toxic metabolites such as 7DHC.
Simvastatin is a known inhibitor of HMG-CoA reductase, and most importantly is able to cross the blood–brain barrier. It has been reported to decrease the levels of 7DHC, as well as increase the levels of cholesterol.
The increased cholesterol levels are due to simvastatin's effect on the expression of different genes. Simvastatin increases the expression of DHCR7, likely leading to increased activity of DHCR7.
It has also been shown to increase the expression of other genes involved in cholesterol synthesis and uptake. However, these benefits are dependent on the amount of residual cholesterol synthesis. Because some individuals possess less severe mutations and demonstrate some amount of DCHR7 activity, these people benefit the most from simvastatin therapy as they still have a partially functioning enzyme.
For individuals that show no residual DCHR7 activity, such as those homozygous for null alleles or mutations, simvastatin therapy may actually be toxic. This highlights the importance of identifying the specific genotype of the SLOS patient before administering treatment. It is still unknown if simvastatin will improve the behavioural or learning deficits in SLOS.
Antioxidant supplementation
Antioxidants are those which inhibit the oxidation of molecules or reduce metabolites that were previously oxidized. Given that some symptoms of SLOS are thought to result from the peroxidation of 7DHC and its derivatives, inhibiting this peroxidation would likely have beneficial effects.
Antioxidants have been shown to increase the level of lipid transcripts in SLOS cells, these transcripts play a role in lipid (cholesterol) biosynthesis and are known to be down-regulated in SLOS.
Furthermore, vitamin E specifically is known to decrease DHCEO levels, which is an indicator of oxidative stress in SLOS, as well as present beneficial changes in gene expression. Vitamin E appears to be the most powerful antioxidant for treating SLOS, and in mouse models has reduced the levels of oxysterols in the brain.
However, antioxidants have only been studied in animal models of SLOS or isolated SLOS cells. Thus, their clinical significance and negative side effects are still unknown, and their use has yet to be studied in humans.
Further considerations
When treating SLOS, a recurring issue is whether or not the intellectual and behavioural deficits are due to fixed developmental problems (i.e. fixed brain malformations), or due to ongoing abnormal sterol levels that interrupt the normal function of the brain and other tissues.
If the latter is true, then treatments that change the sterol levels and ratios, particularly in the brain, will likely improve the developmental outcome of the patient. However, if the former is true, then treatment is likely to help only with symptoms and not with specific developmental deficits.
As mentioned, no treatment has so far proven effective long-term for patients with Smith-Lemli-Opitz syndrome. Potentially, cholesterol supplementation is a logical treatment because it may be expected to raise plasma and tissue cholesterol levels.
By feedback inhibition of hydroxymethylglutaryl-coenzyme-A-reductase, cholesterol supplementation may reduce levels of 7-DHC and related cholesterol intermediates that may be toxic.
Some suggestion of a potential therapeutic response has been seen with hydroxymethylglutaryl-coenzyme-A-reductase inhibitors (statins), but adverse events are not infrequent.
Related Disorders Associated with Smith Lemli Opitz Syndrome
Meckel syndrome is an autosomal recessive disorder. Meckel syndrome is caused by 13 different genes. It affects 1 in every 13,250 and 1 in every 140,000 persons. Babies with Meckel syndrome, like those with SLOS, may have additional toes or fingers (polydactyly).
They often have big cystic (polycystic) kidneys. It is possible for the spinal cord and brain to grow incorrectly (neural tube abnormalities). The heart, facial features, urinary system, bones, eyes, and genitals may all be affected by Meckel syndrome. Meckel syndrome, like SLOS, differs from person to person.
A lack of squalene synthase leads to problems producing cholesterol. It's an autosomal recessive disease caused by mutations in the FDFT1 gene. The symptoms and indicators are quite similar to SLOS. Squalene synthase insufficiency, on the other hand, is far more uncommon.
This ailment has only been recorded in three people. There is no 7-DHC accumulation in squalene synthase deficiency.
MEND syndrome, Dubowitz syndrome, Nguyen syndrome, pseudo-trisomy, Noonan syndrome, Pallister-Hall syndrome, Desmosterolosis, and lathosterolosis are all similar diseases.
Other Names for Smith Lemli Opitz Syndrome
Alternative names for this condition include the following:
- SLO syndrome
- 7-dehydrocholesterol reductase deficiency
- SLOS
- RSH Syndrome
Conclusion
SLOS is a hereditary disorder that affects many organs. Changes in the DHCR7 gene produce this autosomal recessive genetic disorder. SLOS problems are frequently obvious before or soon after delivery (congenital).
SLOS is characterized by sluggish development, a tiny head (microcephaly), and moderate to severe intellectual impairment. Extra toes and fingers (polydactyly) are also frequent birth abnormalities. SLOS is a condition that varies. Some people with SLOS exhibit normal growth and very slight symptoms.
SLOS affects one in every 20,000 to one in every 60,000 newborns born. People with SLOS are unable to produce cholesterol. Extra cholesterol treatment may assist with certain SLOS symptoms, but there is presently no cure for SLOS.
All right, guys, that is it for now for Smith-Lemli-Opitz syndrome. I hope Healthsoothe answered any questions you had concerning Smith-Lemli-Opitz syndrome.
Feel free to contact us at contact@healthsoothe.com if you have further questions to ask or if there’s anything you want to contribute or correct to this article. And don’t worry, Healthsoothe doesn’t bite.
You can always check our FAQs section below to know more about Smith-Lemli-Opitz syndrome. And always remember that Healthsoothe is one of the best health sites out there that genuinely cares for you. So, anytime, you need trustworthy answers to any of your health-related questions, come straight to us, and we will solve your problem(s) for you.
Frequently Asked Questions Associated with Smith-Lemli-Opitz syndrome
What is the Life Expectancy of Someone with Smith-Lemli-Opitz syndrome?
The good news is that, if Smith Lemli Opitz syndrome is properly managed and adequate medical care delivered, those with the condition have the potential to have a normal life expectancy. That said, independent living is unlikely due to the severe intellectual disability that often accompanies this syndrome.
What Does it Mean to be a Carrier of Smith-Lemli-Opitz syndrome?
Smith-Lemli-Opitz syndrome is an autosomal recessive disease caused by mutations in the DHCR7 gene. An individual who inherits one copy of a DHCR7 gene mutation is a carrier and is not expected to have related health problems.
How Many People are Carriers for Smith-Lemli-Opitz syndrome?
Smith–Lemli–Opitz syndrome disease incidence has been studied, primarily in Europe and Canada. Diagnoses have been confirmed by molecular and biochemical methods. Most figures range from 1/60 00011, 12 to 1/20 000.
I Am odudu abasi a top-notch and experienced freelance writer, virtual assistant, graphics designer and a computer techie who is adept in content writing, copywriting, article writing, academic writing, journal writing, blog posts, seminar presentations, SEO contents, proofreading, plagiarism/AI checking, editing webpage contents/write-ups and WordPress management.
My work mantra is: “I can, and I will”
Additional resources and citations
- 1https://medlineplus.gov/genetics/condition/smith-lemli-opitz-syndrome/
- 2https://emedicine.medscape.com/article/949125-overview
- 3https://en.wikipedia.org/wiki/Smith–Lemli–Opitz_syndrome#Treatment

The content is intended to augment, not replace, information provided by your clinician. It is not intended nor implied to be a substitute for professional medical advice. Reading this information does not create or replace a doctor-patient relationship or consultation. If required, please contact your doctor or other health care provider to assist you to interpret any of this information, or in applying the information to your individual needs.